
Research Overview
The research in the Brown Lab involves the application of computational molecular modeling to relate the structure and dynamics of molecular systems to function. We use computational methods to design enzymes, with our strategy being to alter the substrate specificity of existing enzymes. We like to apply all kinds of best practices in molecular modeling to any kind of question - so if you have questions, please contact us through email or our Contact page.
Gallery of Images
PRESENT RESEARCH:
Amyloid βeta-Peptide (Team Amyloid)
IAPP and Small Molecules (Team Amyloid)
HIV (Team HIV)
RNA Virus Drug Discovery (Team RNA Virus)
Protein-Protein Interactions (Team PD-1/PDL-1)
Ribosomal Drug Discovery (Team Ribosome)
PAST RESEARCH:
Radical SAM and Ancient Metabolism (Team Ancient Metabolism)
OR2T7 (Team OR2T7)
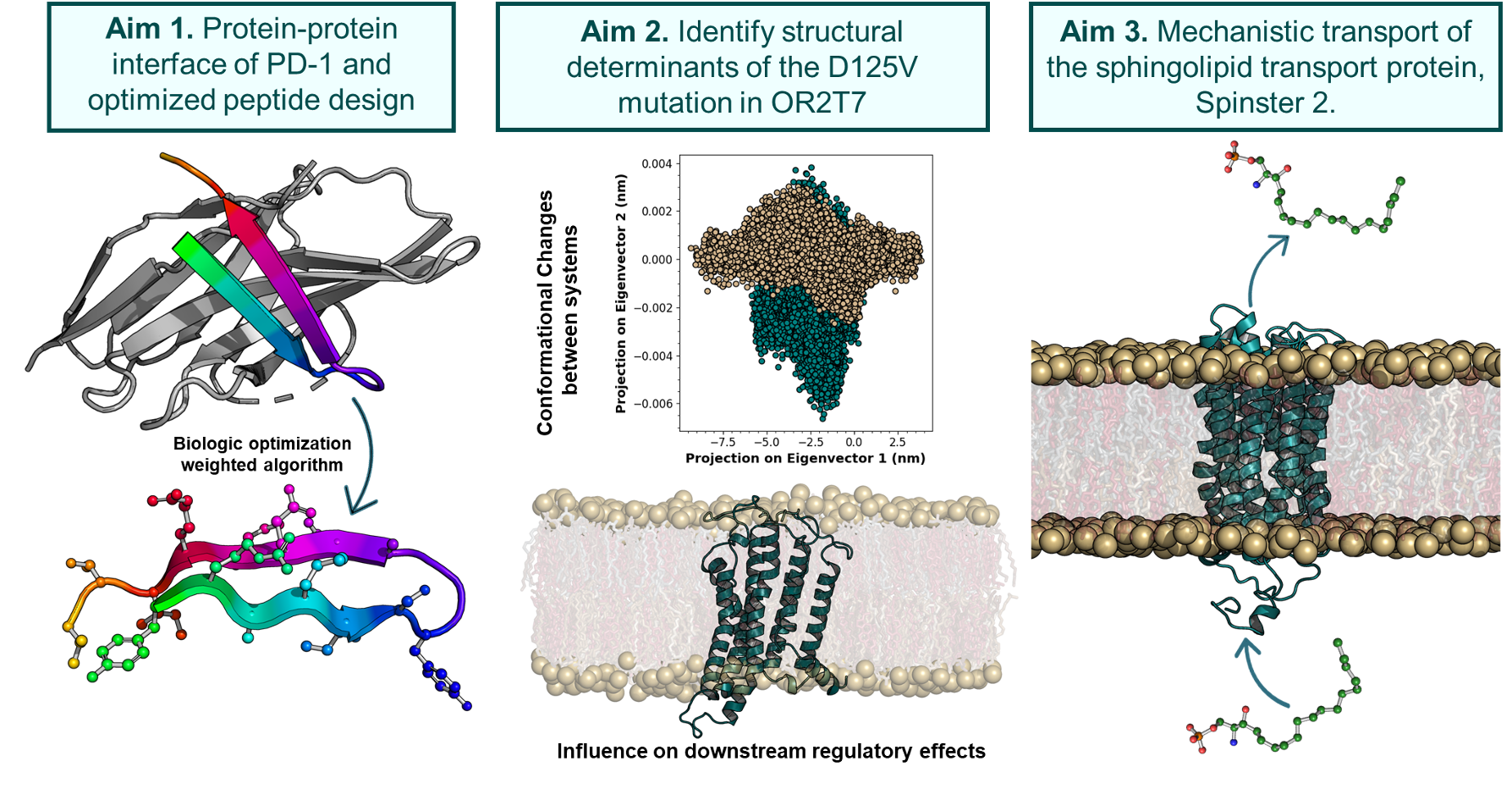
Sphingosine Kinase (Team SPHK)
Peroxisome Proliferator-Activated Receptor (Team PPAR)
IDPs (Team IDP)
Enzymes as Biocatalysts (Team Biochatalysts)
Some of our favorite images that represent the work we do:

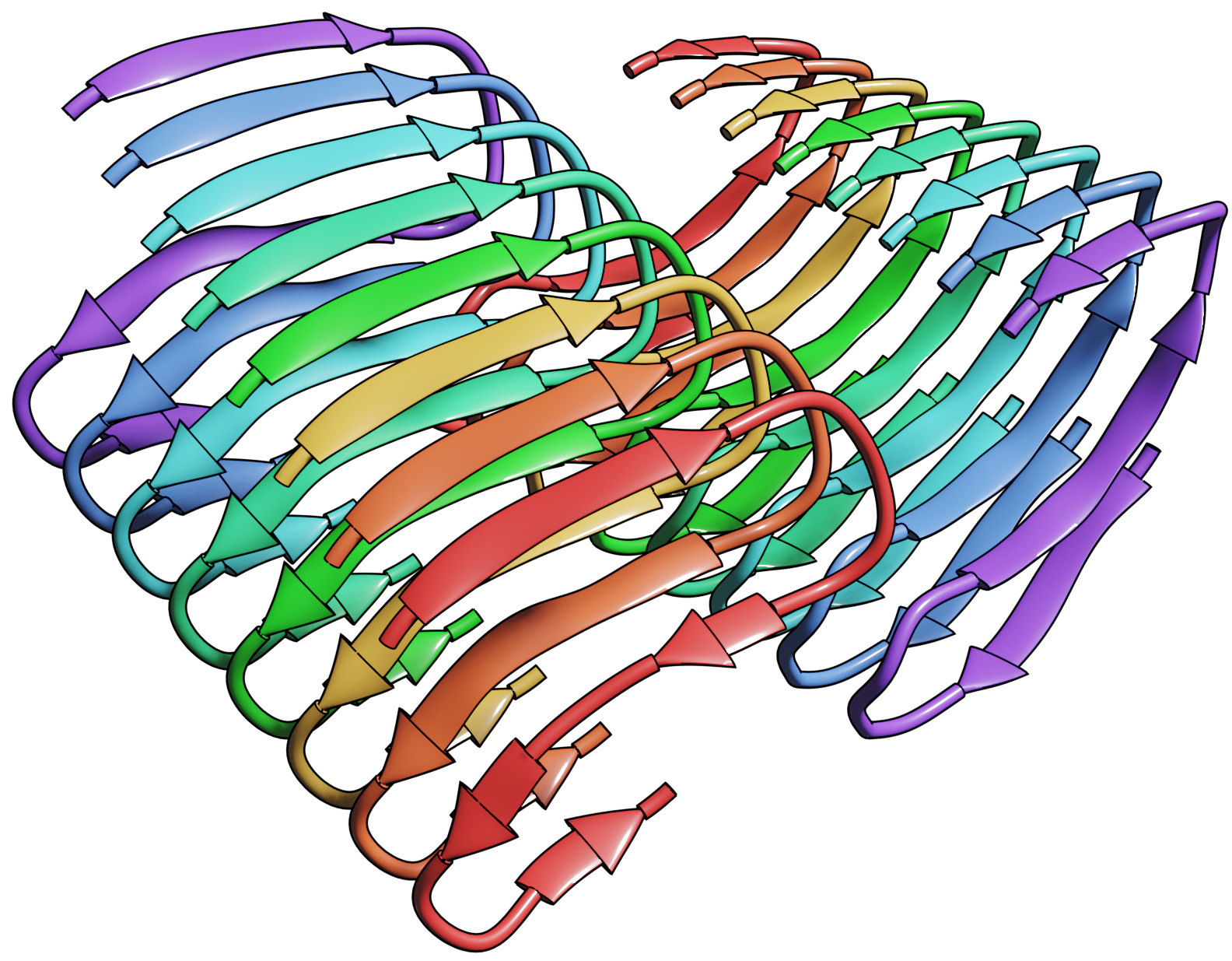
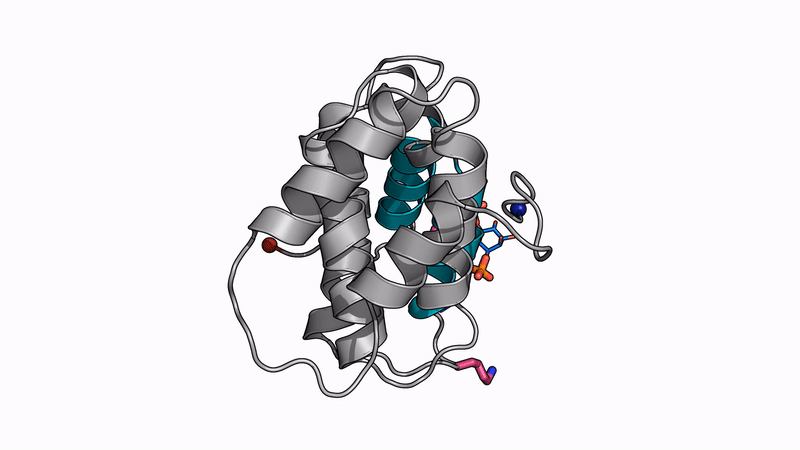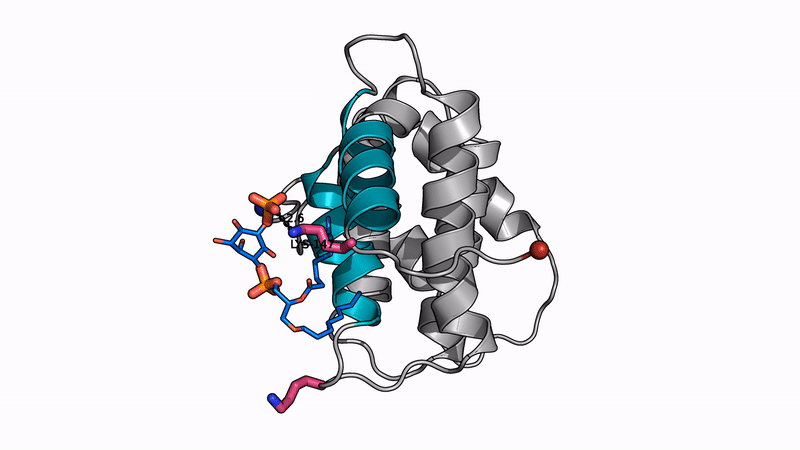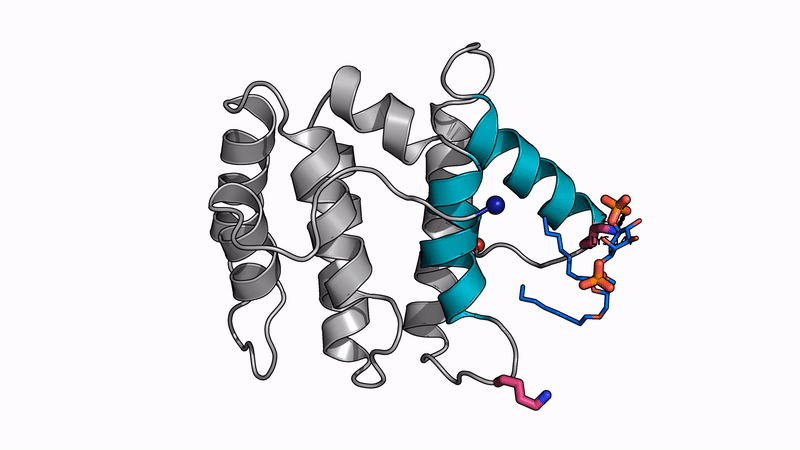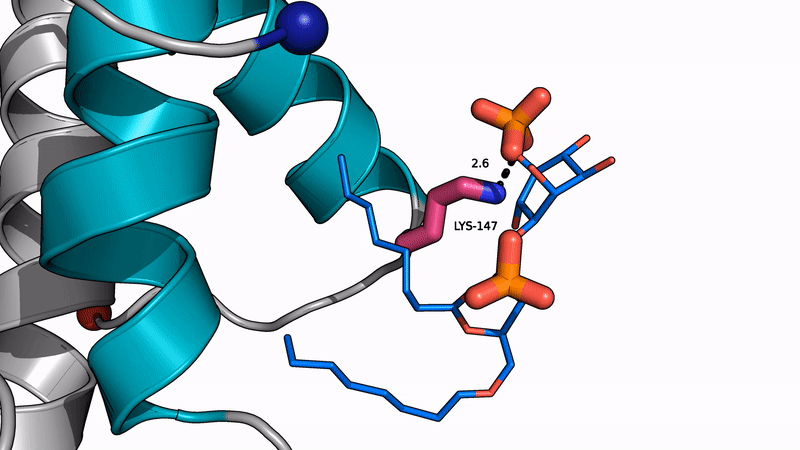
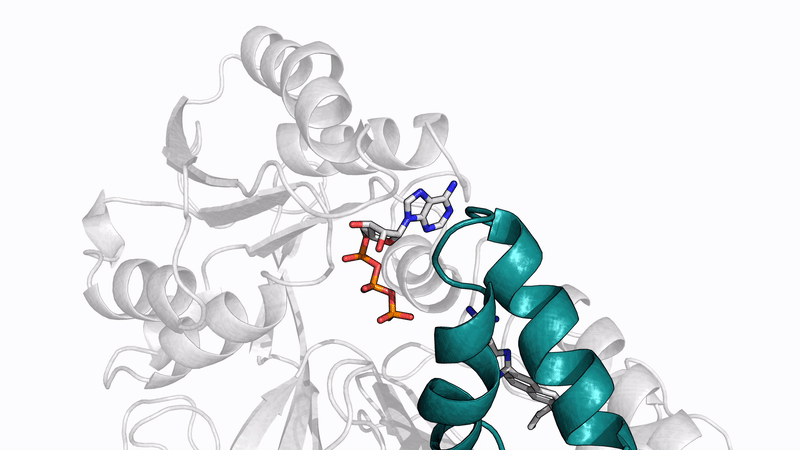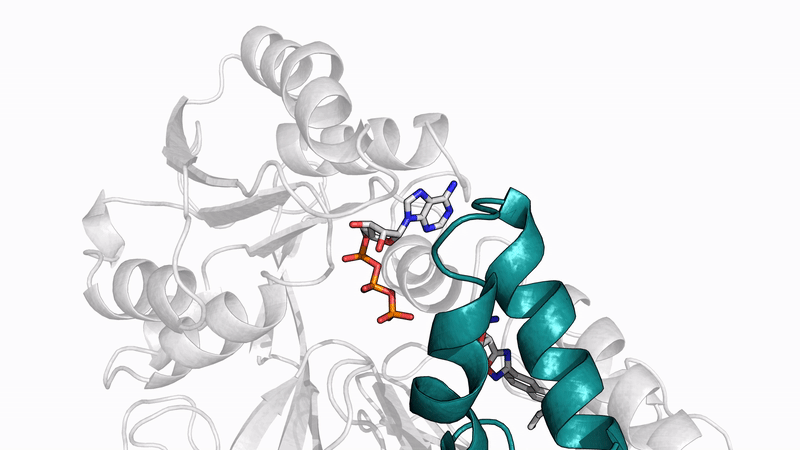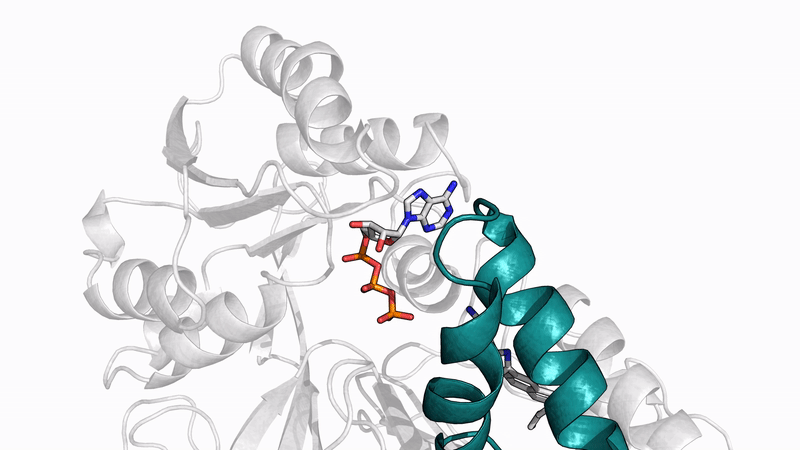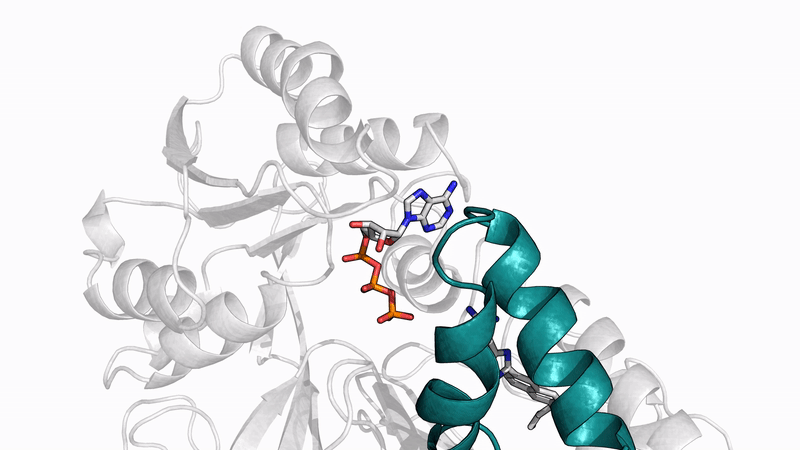

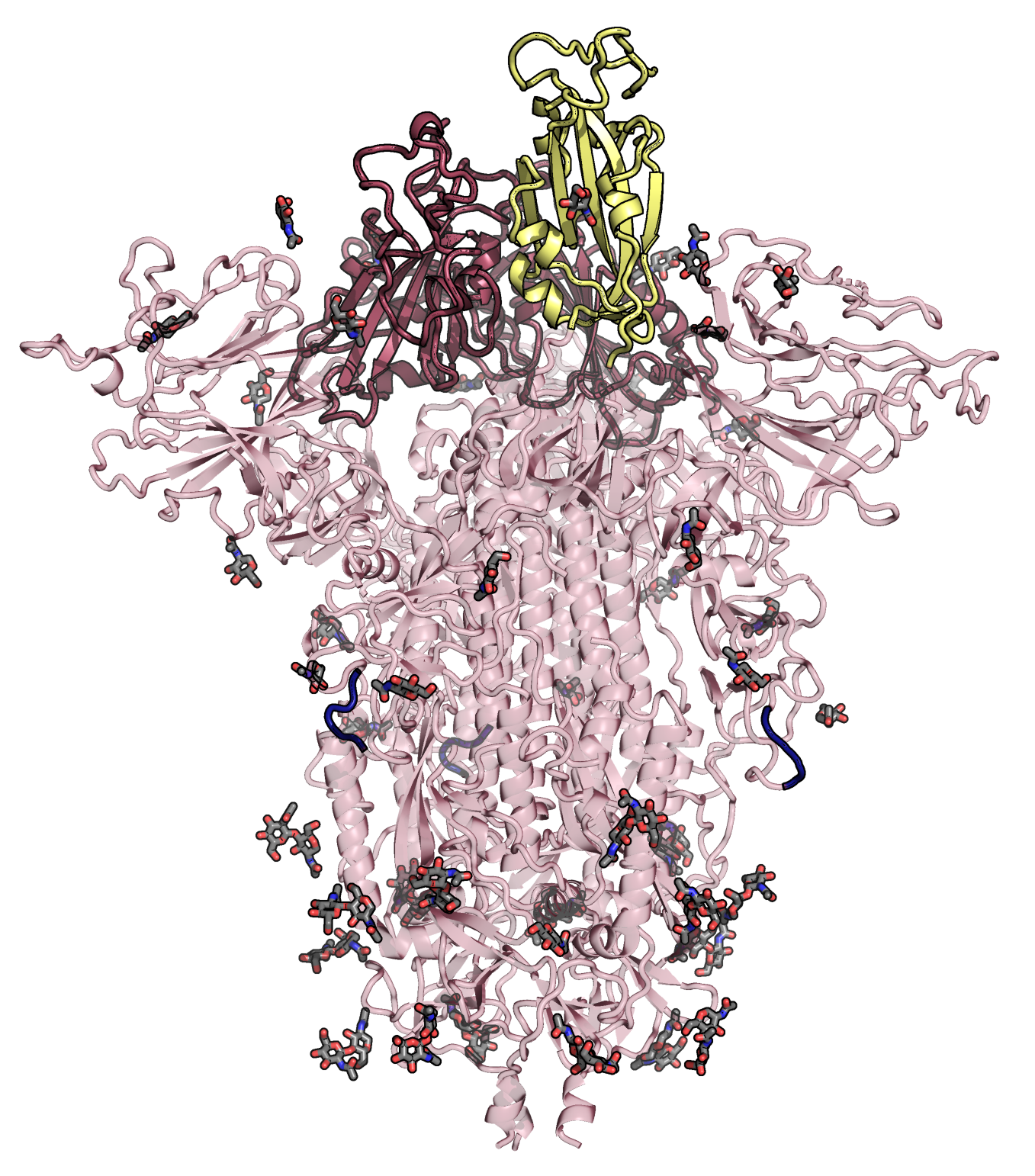
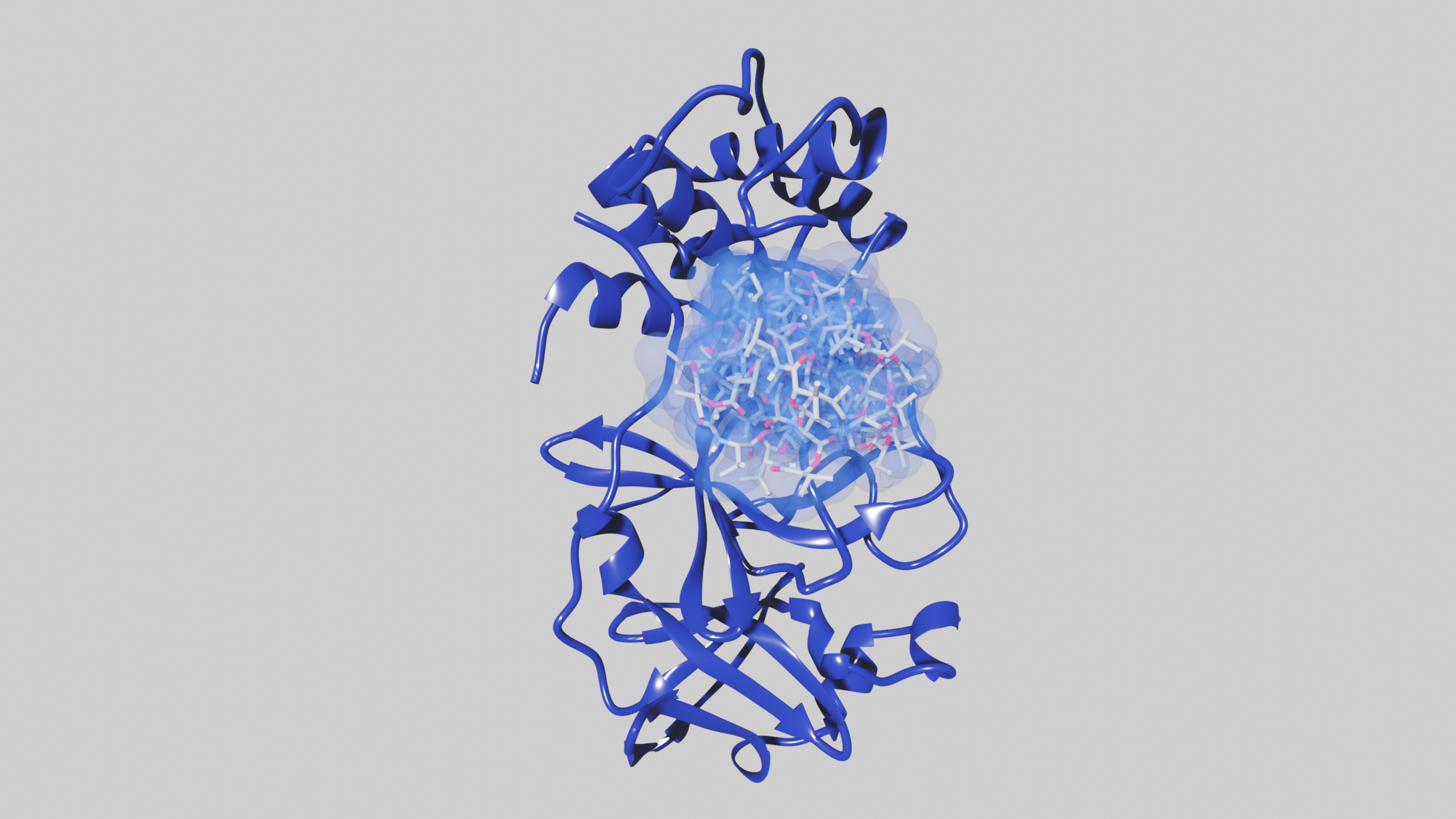
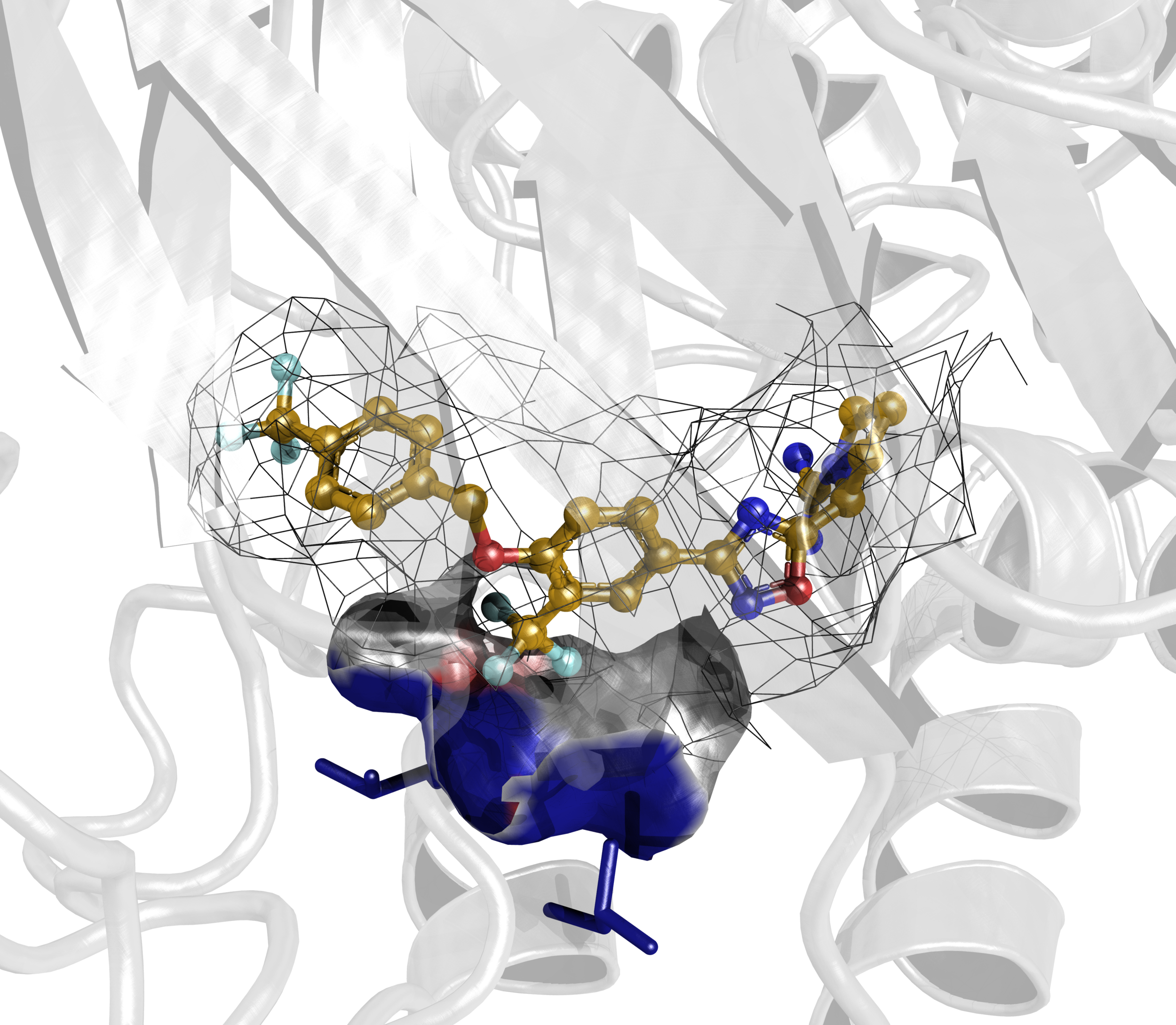

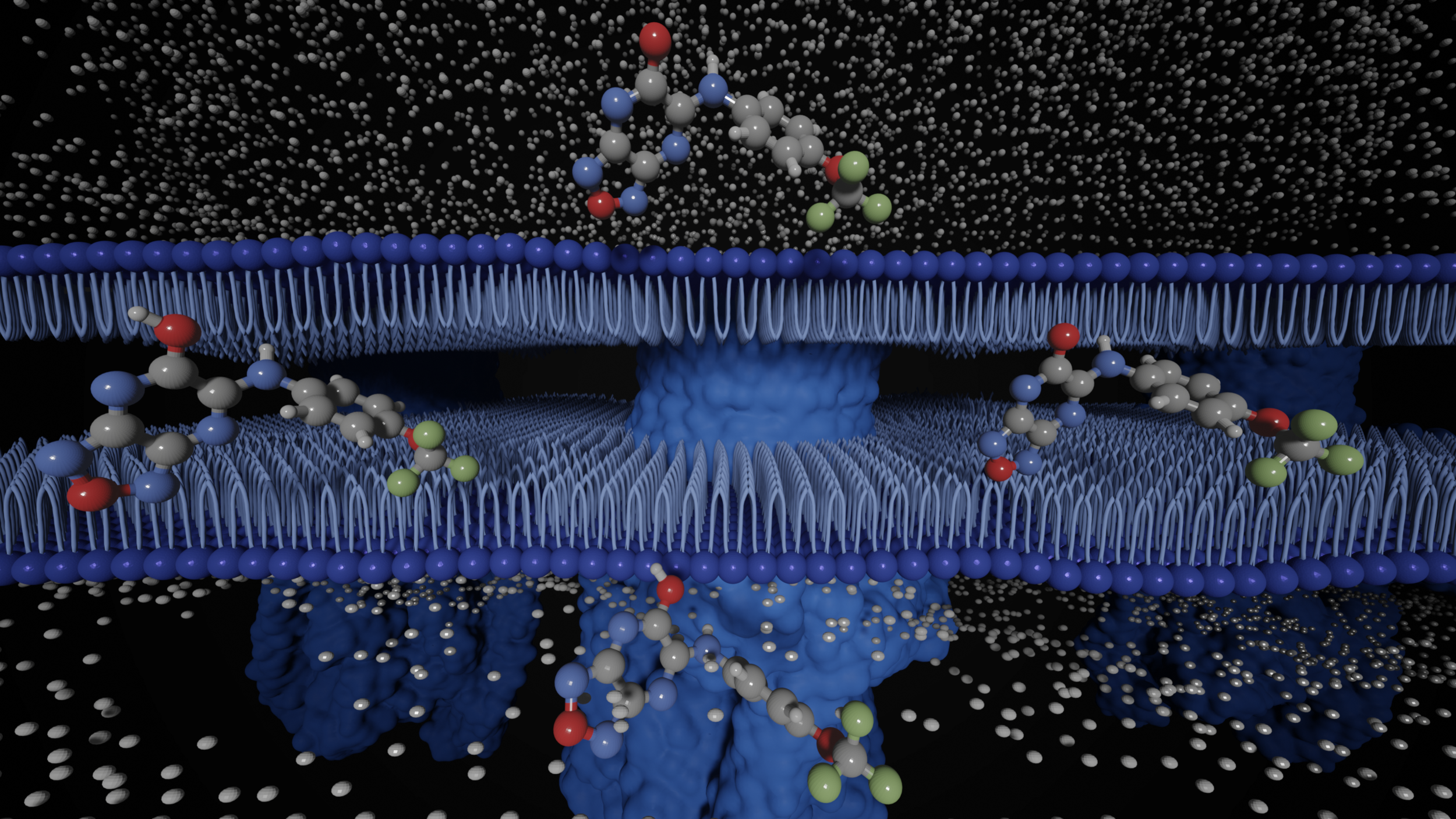
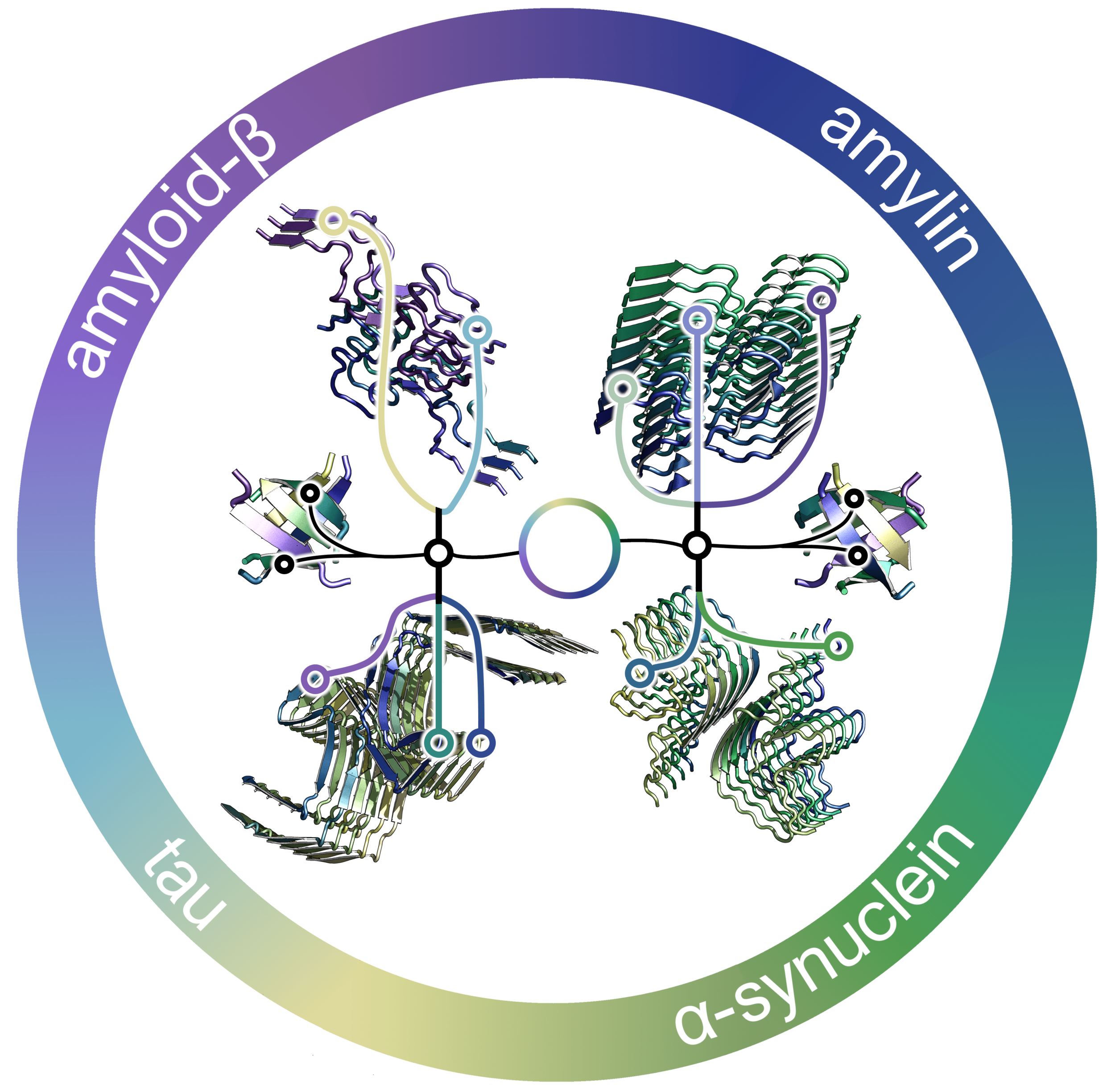
Amyloid βeta-Peptide (Team Amyloid)
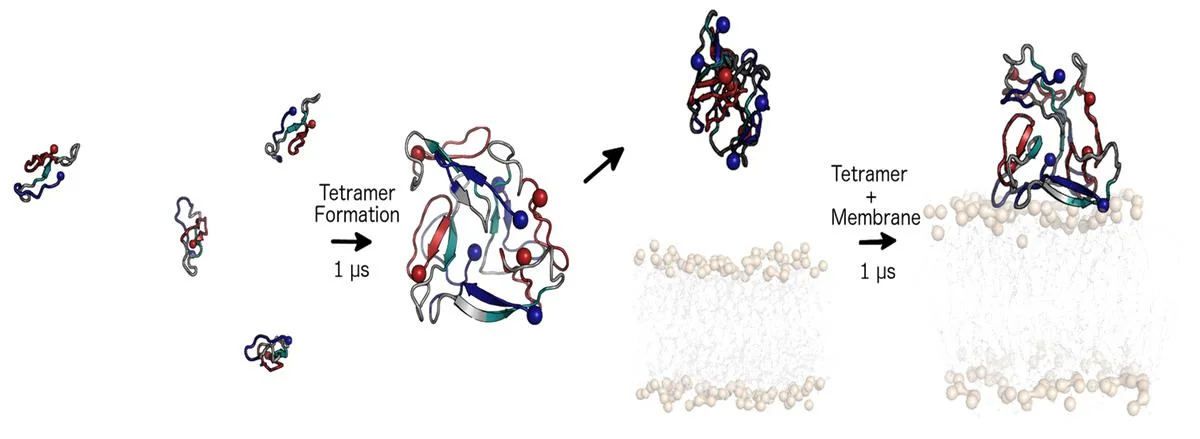
The specific mechanism of toxicity of amyloid beta (Aβ40 or Aβ42) in Alzheimer’s disease (AD) is not well-understood. Two principal theories of Aβ toxicity relevant to neuronal cell death center around the induction of proinflammatory and proapoptotic signals via cell-surface receptor activation or via direct cell membrane disruption. Preventing the accumulation and oligomerization of Aβ is a key potential therapeutic approach for AD and necessary for more biochemical understanding of the disease. We are pursuing three routes, using molecular dynamics (MD) simulations, to better understand the accumulation and oligomerization pathways of Aβ relevant to membrane environments. First, we are using MD simulations, incorporating four full length Aβ42 peptides separated by at least 3 nm, in cholesterol-rich raft model membrane to understand the effect of a membrane environment on the oligomerization of full-length Aβ42, to complement our recent work published in Biophysical Journal. Second, MD simulations are being used to show the formation and structure of n-mer Aβ-42 systems, from dimer to dodecamer, and are expected to demonstrate a gradual shift from beta- barrel-type structures in low n-mer systems to fibrillic or prefibrillic forms in the higher n-mer systems. Third, we are using steered MD to assess the stability of an cylindrical tetramer, formed in previous 1-us simulations, to pull a peptide unit from the oligomer to assess free energy required to disrupt this unit. This approach provides a transitional link between the insoluble fibrillic forms of Aβ and the soluble oligomers and may provide insight into the relative toxicity of Aβ oligomeric species and the contribution of their membrane- and lipid-raft mediated toxicity to the overall cytotoxicity of Aβ. We look to expand our work in this area by better understanding larger low-molecular weight oligomer formation and interaction with receptors, building complex membrane environments to assess other factors, and looking into the role small molecules might play in attenuating some of these membrane disrupting events.
IAPP & Small Molecules (Team Amyloid)
Type 2 diabetes mellitus (T2D) is a complex metabolic disorder drastically increasing in prevalence worldwide. A characteristic feature of T2D is the deposition of islet amyloid polypeptide (IAPP), an intrinsically disordered protein (IDP), in the pancreatic islets of Langerhans, which leads to β-cell dysfunction and death. Interestingly, the rat (Rattus norvegicus) form of IAPP (rIAPP) does not exhibit the toxicity that is observed with the human form (hIAPP). We seek to understand how peptide structure, the presence of flavonoids, and membrane environments influence the folding and aggregation of this protein, which are related to its toxicity. We are using molecular dynamics (MD) simulations to study the influence of primary sequence on secondary and tertiary structures of hIAPP and rIAPP in solution and in the presence of three model membranes. In studies with flavonoids, we use MD to simulate the aggregation of peptide residues 10-20 and 20-29 and examine the efficacy of selected flavonoids on attenuating trimer formation.
HIV (Team HIV)
HIV-1 is a virus that infiltrates many immune cells (T-cells, macrophages, dendritic cells, etc.) and uses them to replicate itself to the detriment of the cell and host body. HIV-1 infects a cell by fusing the viral membrane with the cell membrane, thus releasing a cascade of proteins and genetic material into the cell, which drives its replication. Understanding the mechanisms of viral fusion will be beneficial in possibly providing one route to develop a treatment preventing viral fusion and replication. The primary driver of viral fusion is envelope glycoproteins (gp41), which is located alongside a surface glycoprotein (gp120) on the outside HIV-1 membrane. Gp41 drives viral fusion by invaginating into the host cell and pulling back on itself. Gp41 is comprised of a three-helix structure starting from the C terminus (R660-R683), and a conserved membrane-proximal external region (MPER) (R684-R710) which continues until the N terminus. Some of the most important residues that stabilize the structure of gp41 are several tightly bound arginine groups. These groups have been shown in previous molecular dynamics (MD) simulations to attract water molecules, which helps stabilize the three-helix structure. However, the structure of the MPER has only recently been released, and it is unknown how the addition of the MPER to the three-helix structure will affect those tightly bound arginine groups in a membrane bound solution. MD will be used to see how the addition of the MPER residues will affect the structure of gp41 in a membrane bound solution.
RNA Virus Drug Discovery (Team RNA-Viruses)
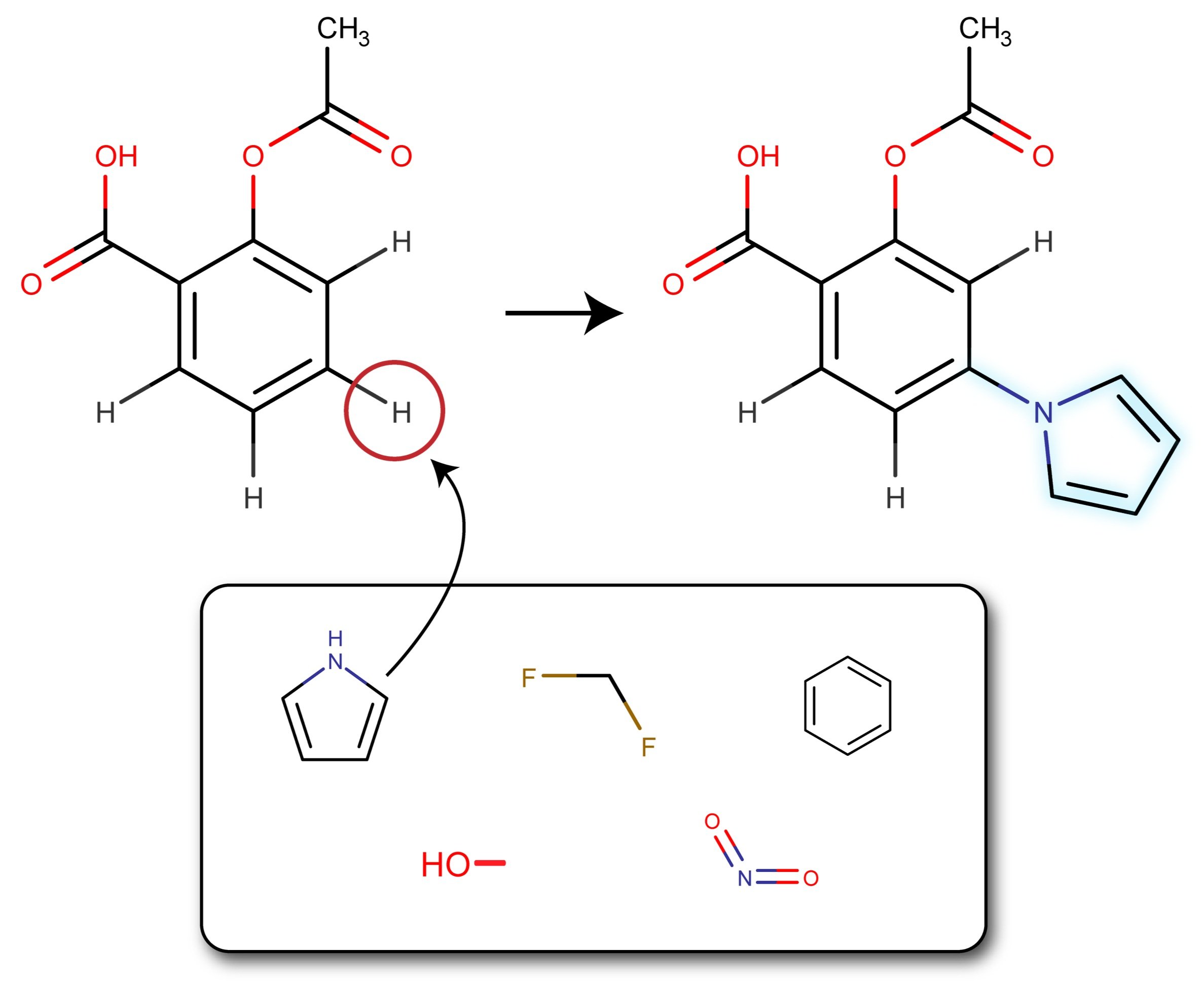
RNA viruses are responsible for several endemics and pandemics, killing millions of people annually. There is a critical need for more efficient drug development methods to combat these viruses. Our lab is investigating a four-stage drug discovery approach. The first stage involves utilizing a drug database to perform high-throughput virtual screening, identifying leads based on energy calculations, residue interactions, and ADME property prediction. The second stage involves late-stage functionalization (LSF) of the top drug leads using a novel framework that combines molecular docking with evolutionary algorithms. Once these drugs are developed computationally, the third stage involves chemically synthesizing these top drug leads and the last stage involves validating the functional activity and mechanistic insight in vivo and by pharmacophore-model-based modeling. To validate this framework, the top drug leads will be identified and then functionalized for the proteases in SARS-CoV-2, Dengue, and Chikungunya RNA viruses and then tested in vivo for efficacy.
Protein-Protein Interactions (PD-1/PDL-1)

Program cell death-1 (PD-1) and its ligand, PD-L1, are validated targets for several cancers that utilize PD-1/PD-L1 to inhibit endogenous anti-tumor immune responses such as those in non-small cell lung cancer (NSCLC). NSCLC accounts for 84% of all lung cancer diagnoses and is associated with a 5-year survival rate of 23%. Lung cancer is also the second most common cancer resulting in greater than 142,000 deaths each year. Identifying the contact regions between a protein and its binding partners is essential for creating therapies that block the interaction. Unfortunately, such contact regions are extremely difficult to characterize because they are hidden inside the binding interface. Protein painting represents a new drug discovery platform that employs small molecules as molecular paints to tightly coat the surface of protein-protein complexes. The molecular paints, which block trypsin cleavage sites, are excluded from the binding interface. Following mass spectrometry, only peptides hidden in the interface emerge as positive hits, revealing the functional contact regions that are drug targets.
Ribosomal drug discovery (TEam ribosome)

Antimicrobial resistance (AMR) to antibiotics is a critical, rapidly increasing, worldwide health concern, contributing to and directly causing millions of deaths annually. Many classes of antibiotic drugs confer inhibitory activity against bacterial infections by binding a single ligand to the bacterial ribosome, typically in the Peptidyl Transferase Center (PTC). However, bacterial ribosomes are adapting to these broad-spectrum treatments more quickly than new antibiotics can be developed. Therefore, new methods of antibiotic research need to be explored. Our team has established, validated, and utilized computational protocols to investigate ribosomal drug discovery. New ways to exploit antibiotic binding patterns are a result of redocking and molecular visualization of ribonucleoprotein complexes. The creation of a linker to join these single ligand drugs would lead to a novel class of antibiotics, bidentates. Bidentate antibiotics demonstrate a unique advantage over single-ligand treatments, as they would be better suited to target specific species of bacteria. Additionally, targeted species would have to develop new adaptations in 2 vital sites of their ribosome, hindering the development of AMR. To determine viable antibiotic candidates for derivatization and linking, our group employs a synergistic, two-pronged approach combining computational modeling studies and medicinal chemistry. Computational modeling studies give us unique insight into antibiotic binding patterns within the ribosome to develop derivative antibiotics with efficacy higher than the parent compound. The corollary synthesis and testing of these derivatives by the medicinal chemistry prong have shown our in-silico predictions of drug efficacy have reached 71% accuracy. This achievement in and of itself may lead to significantly reduced antibiotic production turnaround and cost.
Past Projects
Radical SAM and Ancient Metabolism (Team Ancient Metabolism)

Radical S-adenosyl-L-methionine (rSAM) proteins are a superfamily of enzymes that contain 4Fe-4S clusters, usually coordinated by a vital CXXXCXXCX moiety. The 4Fe-4S clusters allow the enzymes to do radical chemistry by cleaving the substrate, SAM, to produce a free electron. This unique ability of radical SAM proteins lets them serve many different roles in living systems. Our group is investigating rSAM enzymes that may be involved in the metabolic pathways for archaeal methanogenesis. Genomic analysis has helped identify DNA sequences as possible rSAM genes in certain archaea. Homology modeling and model validation give insight into the potential structure of these gene products. Molecular visualization of validated homology models indicates if the protein structure is arranged in a manner by which the CXXXCXXCX moiety can effectively coordinate the 4Fe-4S cluster to do radical chemistry.
OR2T7 (Team or2t7)

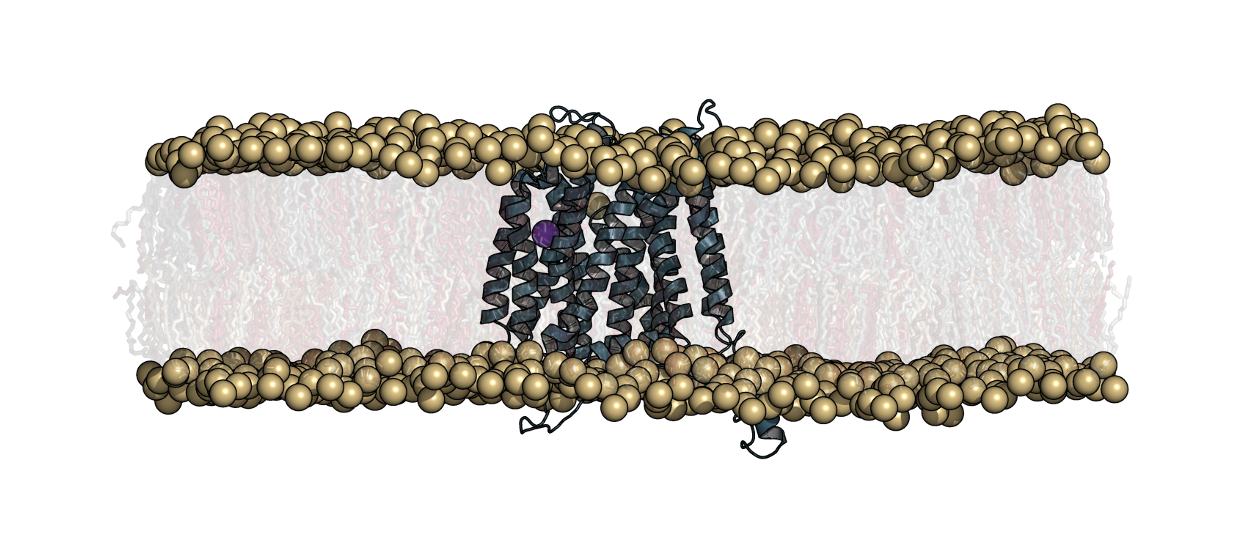
Glioblastoma multiforme (GBM) is one of the most malignant and common type of central nervous system tumors. Surgery, radiation therapy, and chemotherapy are the primary treatment options available, however GBM remains incurable and is one of the most challenging tasks in clinical oncology. Recently, the cancer genome atlas was analyzed and identified a gene, olfactory receptor 2T7 (OR2T7), with a rare somatic mutation (D125V). Not previously studied, OR2T7 could possibly represent a novel target G protein-coupled receptor (GPCR) for treating GBM. The objective of our research is to understand the functional mechanisms of OR2T7 and determine if it could be a potential therapeutic target for GBM. In this work, we use in silico techniques to explore the protein structure function to identify which activating ligand binds extracellularly, the G-protein that binds intracellularly, and determine the downstream signaling components.
Sphingosine Kinase (Team SphK)

Alterations in or disruption of cell proliferation and cell signaling pathways are significant given their connection to multiple disease states including cancers and fibrosis. In our current research we focus on two sphingosine kinase isoforms, sphingosine kinase 1 (SphK1) and sphingosine kinase 2 (SphK2). These enzymes catalyze the synthesis of a signaling molecule, sphingosine-1-phosphate (S1P), and they are involved in cell differentiation and have been found elevated levels in certain cancer cells. This connection has led to an increased focus for more potent inhibitors that are selective for SphK1 or SphK2. Mouse SphKs produce different levels of S1P under certain conditions compared to human SphKs, so it is necessary to determine the structural properties that may result in different S1P expression levels. In collaboration with the Santos Lab in the Chemistry Department at VT we have produced homology models of mouse SphK1 and SphK2, performed molecular docking to identify isoform-specific inhibitors, and analyzed key interactions between the inhibitors and the enzymes in the binding pocket. We also are analyzing binding site volume to compare the four enzymes and elucidate differences based on sequence variation in the binding cavity. This study has the potential to aid in SphK inhibitor design and inform in vivo studies involving mice by highlighting structural distinctions and identifying the role of key residues that cause observable, functional differences in isoforms and species. We also seek to explore more in silico methods (QSAR, COMFA) to best determine key physicochemical properties for isoform specific inhibitors.
Peroxisome Proliferator-Activated Receptor (Team PPAR)
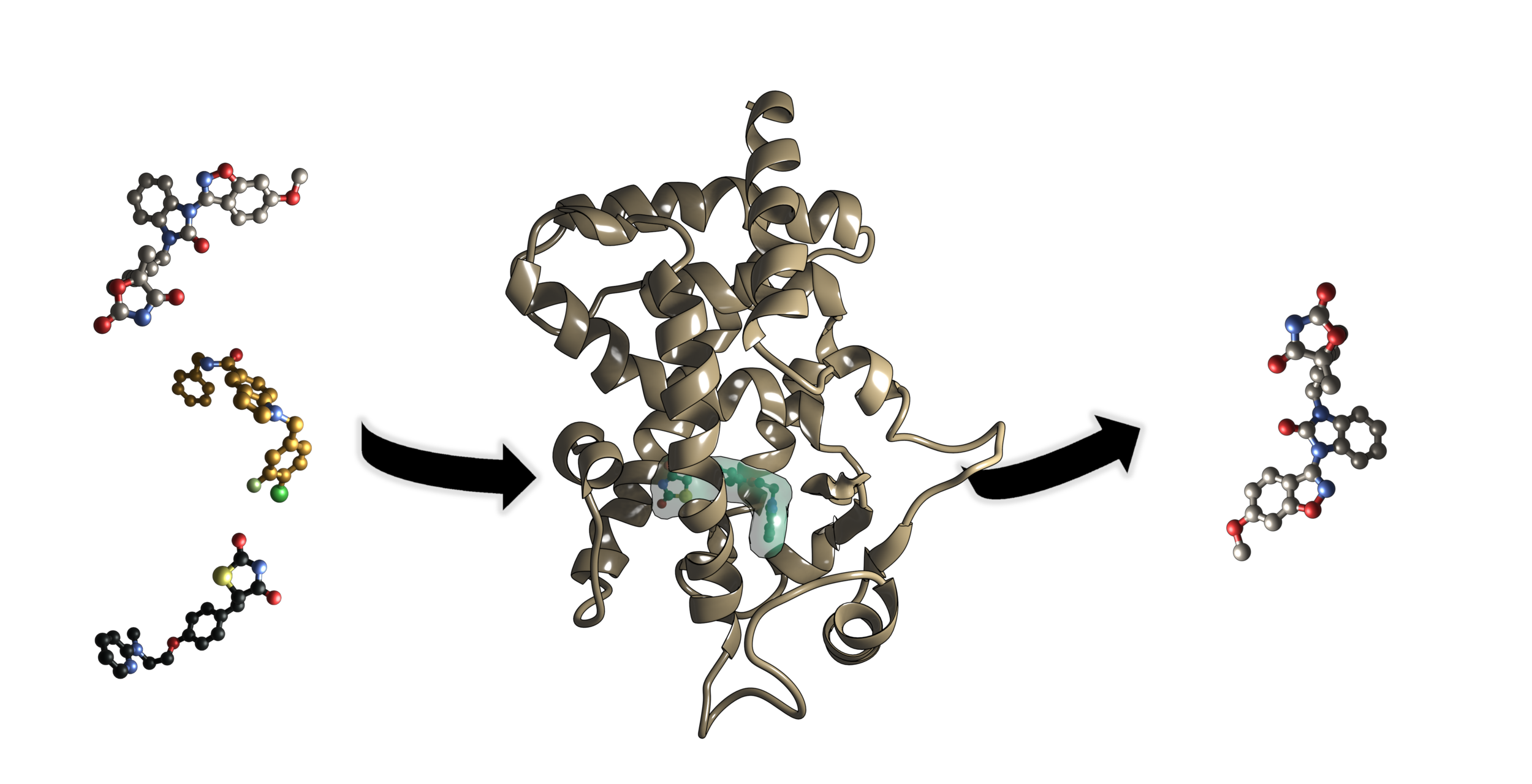
The number of Americans with chronic inflammation-related diseases, including diabetes and obesity, rises significantly each year. The therapeutics used to treat type 2 diabetes, thiazolidinediones (TZDs), often exacerbate the problem by causing side effects, like edema and heart complications. In an attempt to discover more effective and less harmful treatments, our group is working on a combined computational and experimental approach for finding PPARγ, PPARδ, and PPARα agonists. PPARs are in the nuclear receptor family, and transcription is activated and mediated by ligand binding. The molecular composition of the ligand dictates conformational changes that lead to activation of the protein and coactivator recruitment for regulation of genes. Specifically, PPARγ is a key drug target for the regulation of Type II Diabetes and inflammatory diseases due to its role in mediating the levels of glucose and controlling inflammation levels around the body. We are using MD to assess the apo, active, inactive, unbound, and bound structures of PPARγ and the role ligands play in altering the conformational state of PPARγ. Particular interest is in the oscillation of helix 12 (H12), residue fluctuation, and secondary structure composition. The outcome will be a compilation of protein-ligand interactions required for transition of each structure type an active form. This information would benefit drug discovery and design efforts to find alternative treatments for chronic inflammatory disorders.
IDPs (Team IDP)

Intrinsically disordered proteins (IDPs) lack stable tertiary and/or secondary structure under physiological conditions and are often implicated in disease states (Alzheimer's disease, Type 2 Diabetes, Parkinson’s, etc), through the assembly of proteins into oligomeric aggregates. The details of this self- and hetero-assembly and the effects of these oligomers, especially low molecular weight oligomers (dimers to dodecamers), on plasma membranes is largely unknown. By investigating the molecular details, mutations, and interactions associated with oligomer assembly and the interactions of the oligomers with membranes, we can provide insight into mechanistic details relevant to toxicity and disease states. In this work, we seek to use in silico techniques to characterize folding/unfolding of IDPs, including amyloid beta and islet amyloid polypeptide, both separately and in heterogeneous systems (combining multiple IDPs together). We also seek to assess potential efficacy of small molecules to attenuate their oligomerization that leads to fibril formation.
RCAN
RCAN 1 is a protein located on chromosome 21 that is upregulated in down syndrome. People with down syndrome are resistant to certain types of cancer and the mechanism of resistance is thought to be caused by a decrease in angiogenesis around tumor cells. To study this we are trying to better understand the interactions between RCAN1 and RAF1, in the hopes that we can identify key residues and regions of this interaction. This could possibly lead to the development of small molecule inhibitors in the long term. Our main focus now is to computationally characterize this interaction using protein-protein docking and molecular dynamics.
Enzymes as Biocatalysts
We have recently initiated a collaborative project with Dr. Ryan Senger of the Department of Biological Systems Engineering. Dr. Senger has developed an approach to identify metabolic pathways that may lead to the production of novel biofuels and other chemical intermediates using bacterial systems. These systems have the potential to be very specific for the products they produce and are environmentally friendly. One of the challenges is that the enzymes that are involved in these pathways may have low activity towards the substrates on which they need to act. We are applying molecular modeling approaches to model structures of the enzymes, examine their dynamics, dock potential substrates, and predict changes in the enzyme that may improve activity towards the substrates.
Peanut allergens
Computational tools and techniques are attractive due to the low-cost and scope of application and are becoming more prevalent and accepted across a variety of fields. With the advancement of high performance computing resources and the recent utility of GPU architecture, molecular dynamics (MD) simulations have become a nearly ubiquitous tool to interrogate protein structure-function relationships. Understanding these structure-function relationships is fundamental to protein biochemistry and facilitates avenues to modulate activity in disease. Herein, we study and compare the ability of several GPU- and CPU-based clusters to accelerate mechanistic and therapeutic discovery with MD simulations. MD simulations on the peanut protein Ara h 2, the causative agent of allergenicity, from 10 different peanut species of the genus Arachis were conducted in triplicate out to 800 ns to probe epitope solvent accessibility relevant to allergenicity. These simulations provide molecular insight into the causes and potency of the allergenic response. Ultimately, these computational findings and insights can be translated to experimental labs, increasing the impact of the work, understanding of disease states, and guiding therapeutic design. Further, integrating GPUs into these parallel computing operations accelerates computations and provides researchers with tools to perform MD simulations without large CPU centers. We have thus provided an increased understanding of protein structure-function relationships through the usage of high performance computing and have shown GPU scaling power with MD simulations.